Metagenomics
Genome-centric metagenomics is a field occupied with the retrieval of genomes from samples containing a mixture of microbes, such as soil, wastewater, or the gastrointestinal system. Typically, the aim is to study the structure and function of the microbes, often in the dynamic context of their natural environments or by their association with a host (microbiomics). Furthermore, genome-centric metagenomics or microbiomics provides a detailed blueprint of the metabolic potential (genes and metabolic pathways) and is often a prerequisite for studying gene expression patterns (metatranscriptomics or proteomics).
The genomes are bioinformatically extracted from one or more de novo assembled metagenome(s) and a set of samples (dimensions) with varying compositions (e.g., different time points or treatments). Each dimension is used to bin contigs together into separate metagenome-assembled genomes (MAGs) of varying quality. A more contiguous assembly supported by many sample dimensions will result in the retrieval of more high-quality MAGs than a fragmented metagenome with few sample dimensions. At DNASense, we are experts in long-read metagenome sequencing and analysis, forming a solid foundation for obtaining contiguous metagenomes and high-quality MAGs.
For the retrieval of prokaryotic MAGs, we currently recommend using the current Oxford Nanopore long-read chemistry (R10.4.1). This greatly improves the retrieval of high-quality, contiguous MAGs and eliminates GC-, amplification-, and loading biases (accurate abundance estimates). We do not recommend using a short read-only approach (e.g., Element Biosciences). However, for sequencing eukaryotes or targeting retrieval of eukaryotic MAGs, we recommend including short-read data (50x) for polishing the retrieved genomes (hybrid sequencing).
If you are considering doing metagenomics, we encourage you to contact DNASense already during the experimental planning and design, setting the foundation for your project’s most optimal result outcome. We offer access to both short-read (Element Biosciences) and the latest long-read (Oxford Nanopore) DNA sequencing platforms, allowing us to tailor sequencing and bioinformatic workflows according to your specific requirements.
The DNASense team has extensive experience within the fields of metagenomics, and our active involvement in state-of-the-art methods and sequencing platforms (read about it in Nature Methods) ensures that customers obtain valuable insight from our tailored bioinformatic analyses.
We have extracted DNA from all types of low- and high-biomass sample matrices. Our DNA extraction workflows can be customized (using both manual and automatic methods) to accommodate most sample types while minimizing DNA extraction biases in complex communities (see Albertsen et al.) and preserving yield and quality (purity and HMW DNA) to the widest possible extent. Our DNA extraction expertise guarantees the most optimal project outcome and is compatible with short-read (Illumina) and long-read sequencing platforms (e.g., Oxford Nanopore sequencing).
Sample matrices include but are not limited to: Prokaryotes, invertebrates, fungi, salmon, wastewater, aquacultures, soil, oil spills, marine/freshwater samples, eDNA (environmental DNA), bioreactors, tree bark, mangrove and marine sediments, pig/chicken/rat/fish entrails/feces, mining/drill sites, cow rumen, seaweed, oysters, mouthwash, tooth swaps, skin swaps, microbial induced corrosion samples, lung tissue, colon cancer biopsies and liver biopsies.
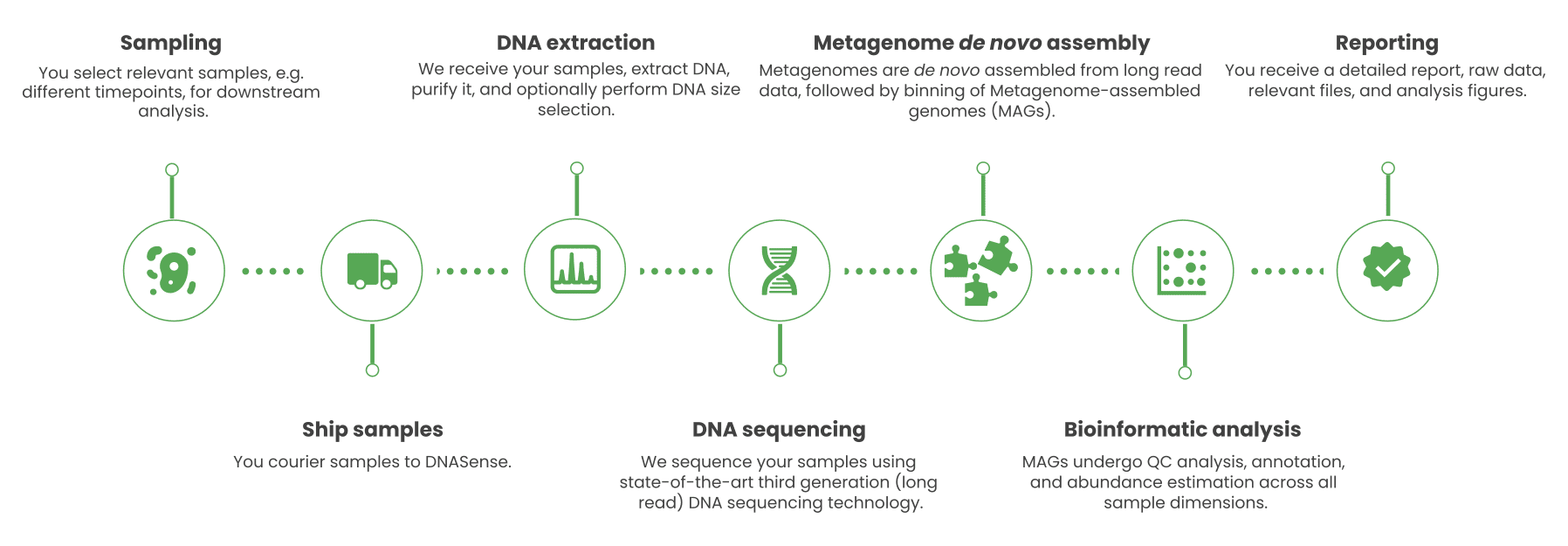
Standard package includes: optional pre- and post-project meetings with a DNASense specialist, DNA extraction, library preparation, sequencing, pre- and post-sequencing quality control, de novo assembly, automatic genome binning, taxonomic profiling, gene annotation, access to raw data, result files and a detailed project report.
Add-on services include but are not limited to: Customized DNA extraction and purification, manual genome binning, Functional annotation (KO, GO, and KEGG), functional enrichment analysis, manual curation of metabolic pathways, gene mining, custom annotation, and data submission.
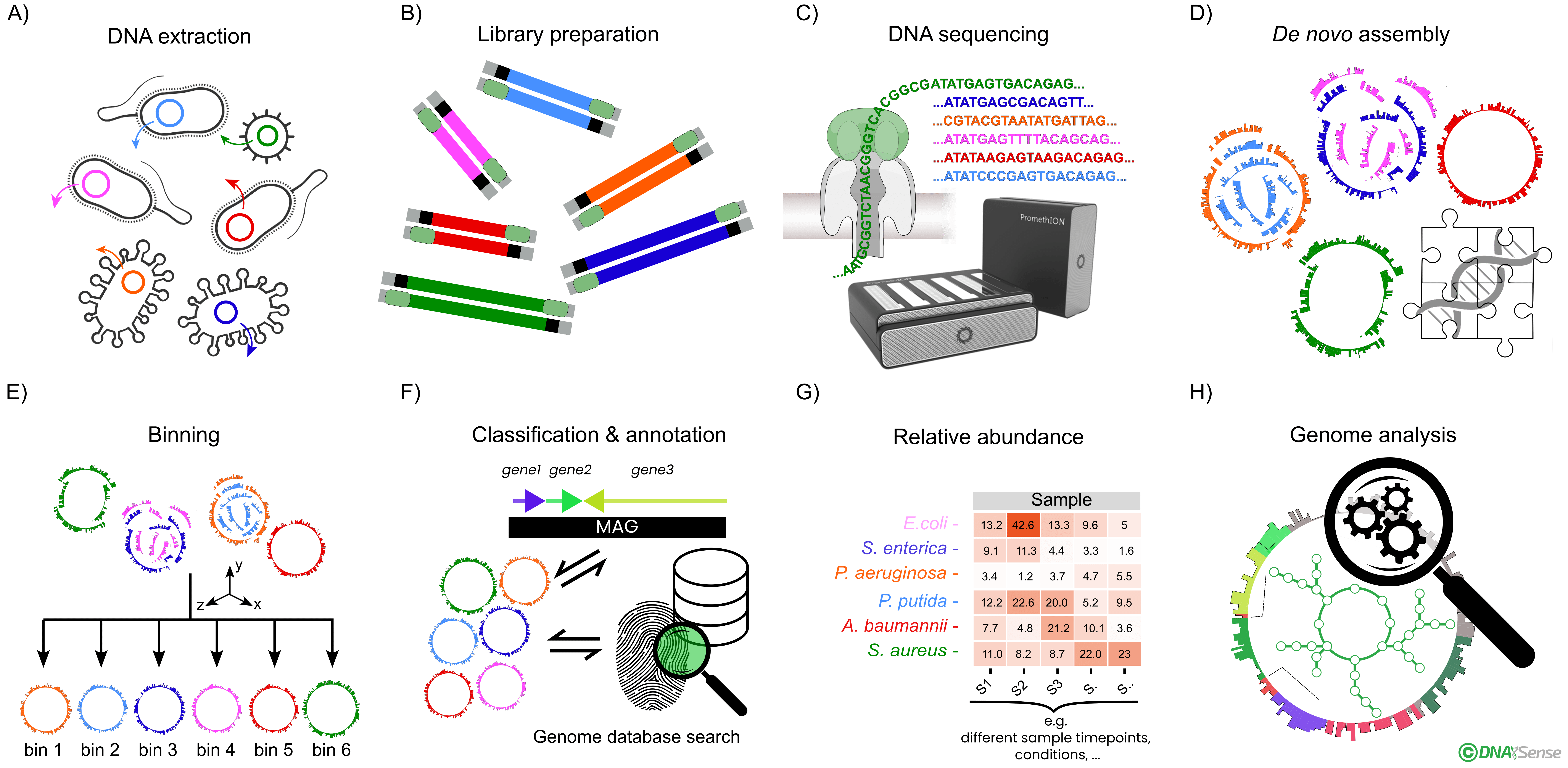
Overview of the general metagenomics analysis workflow. A) Total DNA is extracted from sample biomass, followed by B) native DNA sequencing library preparation and C) DNA sequencing using the Oxford Nanopore Technologies’ platform. D) The obtained DNA reads are quality filtered, and highly contiguous de novo metagenome assemblies are generated. E) Using machine learning models and experiment data variables like time points and treatments, individual metagenome-assembled genomes (MAGs) are isolated into bins. F) MAGs are classified against a state-of-the-art genome and/or rRNA taxonomy database for classification and gene annotation is carried out. G) MAG relative abundance is subsequently estimated. H) High-quality MAGs enable higher order bioinformatic analyses, such as metabolic pathway analysis, functional characterization, and transcriptome profiling.












FAQ
How much sequencing data do I need for metagenome sequencing?
We recommend a minimum sequencing depth of 40-50x, depending on the question. If you want to study a microorganism (5 Mbp genome) present at a 1 % abundance, you would need 20-25 Gbp data.
Is metagenomics suitable for assessing the abundance of very low-abundant organisms?
While it is possible and the less-biased approach, you would need a relatively high sequencing depth. Instead, consider using an amplicon-based approach. It is more sensitive, and you would need less data.
I have heard that Nanopore sequencing is error-prone?
The raw read accuracy of Nanopore sequencing is slightly lower than Illumina, but we use state-of-the-art Q20+ chemistry, which achieves comparable consensus accuracies and handles homopolymers found in prokaryotes (see Nature Methods).
What level of taxonomic resolution can I expect from the classification?
For relatively complete prokaryotic genomes with little or no contamination, we use the Genome Taxonomy Database (GTDB), which potentially provides species-level resolution. Our standard service also includes rDNA extraction and classification against the Silva SSU database (genus level for both prokaryotes and eukaryotes). Custom databases can be included (add-on service).
Why does Nanopore metagenome DNA sequencing reduce composition biases?
Our standard library preparation protocol involves sequencing native DNA. There is no amplification or tagmentation involved.