
Genomics
Whole-genome sequencing, de novo assembly, gene annotation and bioinformatic analysis of pure culture microbial genomes provide detailed insights into functional potential, and are crucial for understanding gene expression patterns (metatranscriptomics and proteomics), strain typing, and comparative genomics.
Long-read DNA sequencing (Oxford Nanopore Technologies) is by far the superior method for reference-grade prokaryote genomes. DNASense also offers short-read DNA sequencing (Element Biosciences) for special use cases, although not recommended for general microbiomics. For some microbial, and especially higher eukaryotes. it is advisable to supplement long-read data with some low-coverage short-read data for final polishing of genome assembly.
The DNASense team has extensive genomics experience and continuously develops cutting-edge bioinformatics and sequencing methods (read about it in Nature Methods), ensuring valuable insights for our customers.

We have extracted DNA from all types of low- and high-biomass sample matrices. Our DNA extraction workflows can be customized (using both manual and automatic methods) to accommodate most sample types while minimizing DNA extraction biases in complex communities (see Albertsen et al.) and preserving yield and quality (purity and HMW DNA) to the widest possible extent. Our DNA extraction expertise guarantees the most optimal project outcome and is compatible with short-read (Illumina) and long-read sequencing platforms (e.g., Oxford Nanopore sequencing).
Sample matrices include but are not limited to: Prokaryotes, invertebrates, fungi, salmon, wastewater, aquacultures, soil, oil spills, marine/freshwater samples, eDNA (environmental DNA), bioreactors, tree bark, mangrove and marine sediments, pig/chicken/rat/fish entrails/feces, mining/drill sites, cow rumen, seaweed, oysters, mouthwash, tooth swaps, skin swaps, microbial induced corrosion samples, lung tissue, colon cancer biopsies and liver biopsies.
Standard package includes: optional pre- and post-project meetings with a DNASense specialist, DNA extraction, library preparation, sequencing, pre- and post-sequencing quality control, de novo assembly, taxonomic profiling, gene annotation, rDNA extraction, access to raw data, result files and a detailed project report.
Add-on services include but are not limited to: Customized DNA extraction and purification, SNV/SV analysis, core genome SNP analysis, core genome MLST analysis, Functional annotation (KO, GO, and KEGG), functional enrichment analysis, manual curation of metabolic pathways, gene mining, custom annotation and data submission.
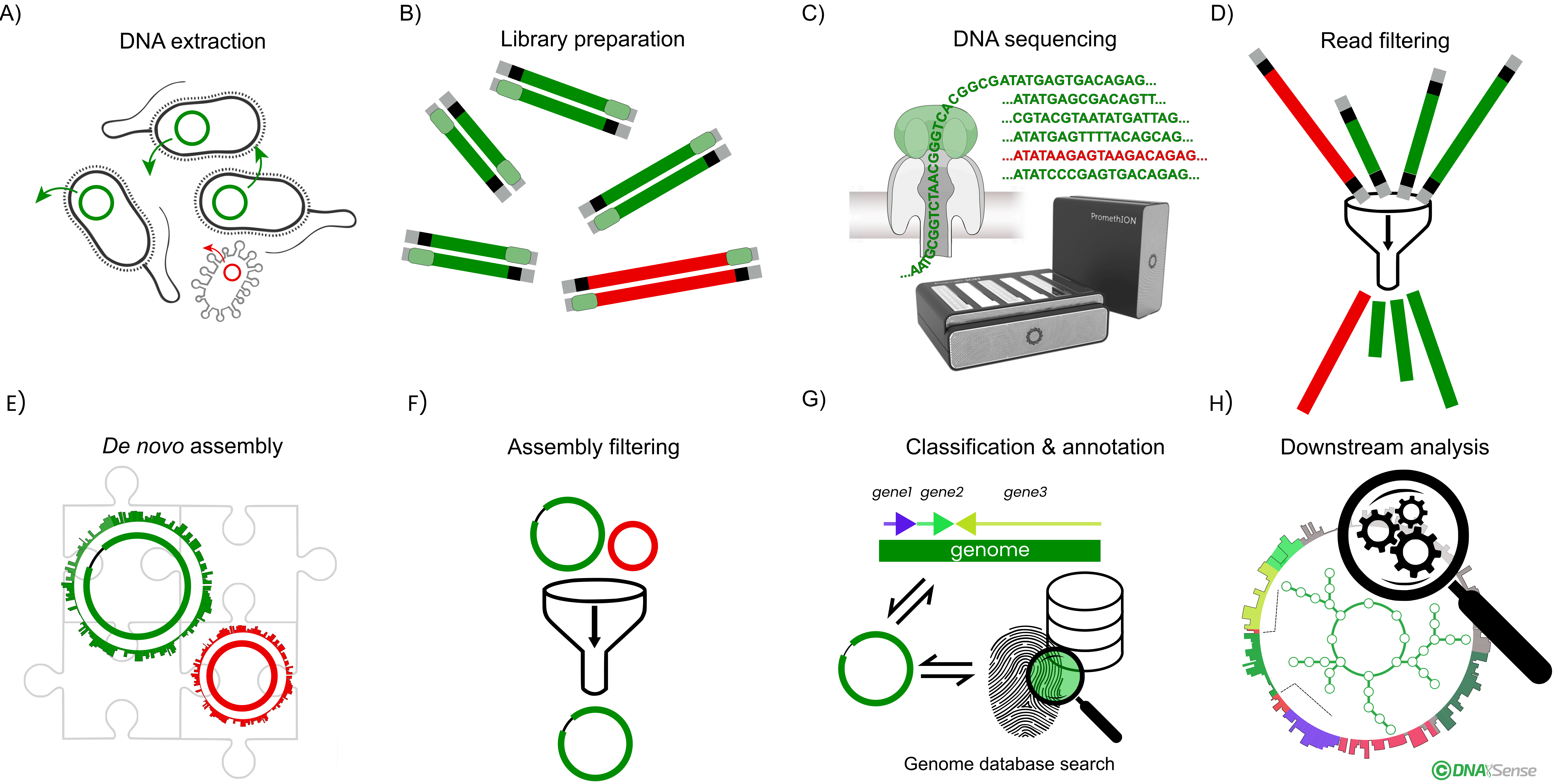
Overview of the general genomics workflow. A) Sample total DNA is extracted from biomass. B) A DNA sequencing library is prepared, and C) sequenced using the Oxford Nanopore Technologies’ platform. D) After quality filtering of the obtained DNA reads, E) de novo genome assemblies are generated. F) Assemblies are subsequently quality-filtered, generating reference-grade assemblies. G) Taxonomy is assigned to each genome by classification against a genome or ribosomal RNA taxonomy database and genes are annotation. H) High-quality genomes enable various bioinformatic analyses, such as metabolic pathway analysis, functional characterization, and transcriptome profiling.












FAQ
Which fast-track options do you offer?
Besides our standard TAT (3 weeks), we offer a fast-track option (7 work days) and an ultrafast-track option (3 work days). Both options are add-ons, and special terms apply.
Should I send biomass or DNA?
We prefer biomass. Extracting high-quality DNA can be challenging and requires that you evaluate the yield (Qubit dsDNA), purity (A260/A280 and A260/A230), and DNA size fragment distribution (e.g., on the Agilent TapeStation using Genomic ScreenTapes).
What level of taxonomic resolution can I expect from the classification?
For relatively complete prokaryotic genomes with little or no contamination, we use the Genome Taxonomy Database (GTDB), which potentially provides species-level resolution. Our standard service also includes rDNA extraction and classification against the Silva SSU database (genus level for both prokaryotes and eukaryotes). Custom databases can be included (add-on service).
How long DNA fragments do I need for long-read sequencing?
It depends on the aim of your analysis. If you wish to produce closed genomes, your DNA read length distribution should be compatible with spanning the longest repeat element in your target genome. For bacteria, this is often the rRNA operon, i.e., reads should be able to span a length of 5000-7000 bp.
I have heard that Nanopore sequencing is error-prone?
The raw read accuracy of Nanopore sequencing is slightly lower than Illumina, but we use state-of-the-art Q20+ chemistry, which achieves comparable consensus accuracies and handles homopolymers found in prokaryotes (see Nature Methods).
Do you have an example report?
Yes. You can request an example report if you wish to see a typical project outcome.
I want to retrieve a genome but my sample is non-axenic. How does this affect things?
If one or more contaminants are present in your sample, we need to adjust the sequencing capacity to match the required depth of the targeted genome. If multiple genomes are present, consider using our metagenomics service.
Case 1: We have extracted DNA from a pure culture bacterium and want to retrieve its 5 Mbp genome at 100x coverage. Therefore, we would need 5 Mbp x 100 x (100/10) = 5000 Mbp.
Case 2: We have extracted DNA from a non-axenic sample and want to retrieve a 5 Mbp genome at 100x coverage. The targeted genome (associated with our organism) is present at 10 % abundance. Therefore, we would need 5 Mbp x 100 x (100/10) = 5000 Mbp.