Metatranscriptomics
Metatranscriptomics and transcriptomics (RNA sequencing) are useful techniques for evaluating the functional activity of whole microbiomes or pure cultures, respectively. DNASense offers RNA sequencing using Element Biosciences’ short-read technology (paired-end 2×150 bp) for gene identification and expression level quantification. These results are further used to identify differentially expressed genes (DEGs) in microbiomes or pure cultures using comparative statistics.
DNASense’s sample-to-answer metatranscriptomics includes advanced bioinformatic pipelines for DEG identification. Our workflows support RNA transcript mapping and identification through de novo assembly, reference genomes, metagenome-assembled genomes (MAGs) or custom genomic references.
A high-quality genomic reference is essential for reference-based RNA-seq. If a genomic reference is not available, one can be generated using genomics or metagenomics. DNASense has extensive experience within the fields of genomics and metagenomics and maintains state-of-the-art bioinformatics and sequencing methods. Importantly, the combination of metagenomics with metatranscriptomics provides a particularly high-resolution analysis optimizing the use of metatranscriptome data and evaluating DEGs at the single MAG level. This leverages both cutting-edge metagenomics and metatranscriptomics for encompassing microbiome analysis.
For large-scale projects, initiating a pilot study is recommended as successful RNA-seq outcomes rely on appropriately scaled experiment designs. The number of biological replicates and sequencing depths can be optimized for the scientific goals, sample complexity, and transcript content. Please consider contacting DNASense during the experiment planning stage for cost-free assistance in optimizing the experiment scale, sampling, and design.
Standard package includes: Optional pre- and post-project meeting with a DNASense specialist, RNA extraction, rRNA depletion, library preparation and cDNA sequencing, Differential Gene Expression (DGE) analysis (mapping/de novo assembly, gene annotation, quantification and statistical analysis), online-access to raw data and result files, a detailed project report with ready to use for publication materials and methods and publication grade illustrations.
Add-on services include but are not limited to: functional annotation (KO, GO), functional enrichment analysis, LEfSe analysis, manual curation of metabolic pathways, Data submission (e.g. NCBI archives).
The above provides general recommendations, but if you are considering to do (meta)transcriptomics, we encourage you to contact DNASense already during the experimental planning and design, setting the foundation for the most optimal result outcome for your project.
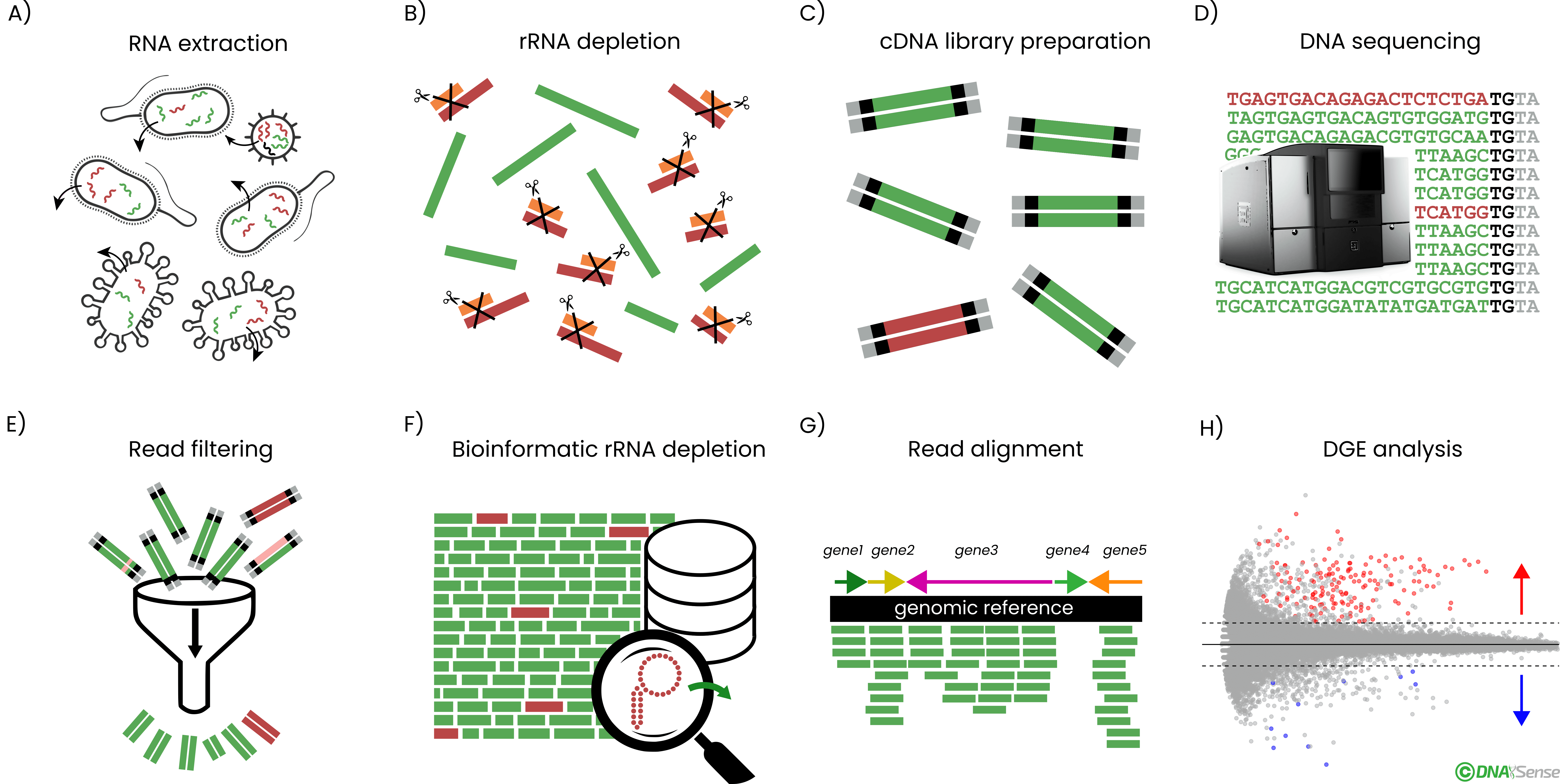
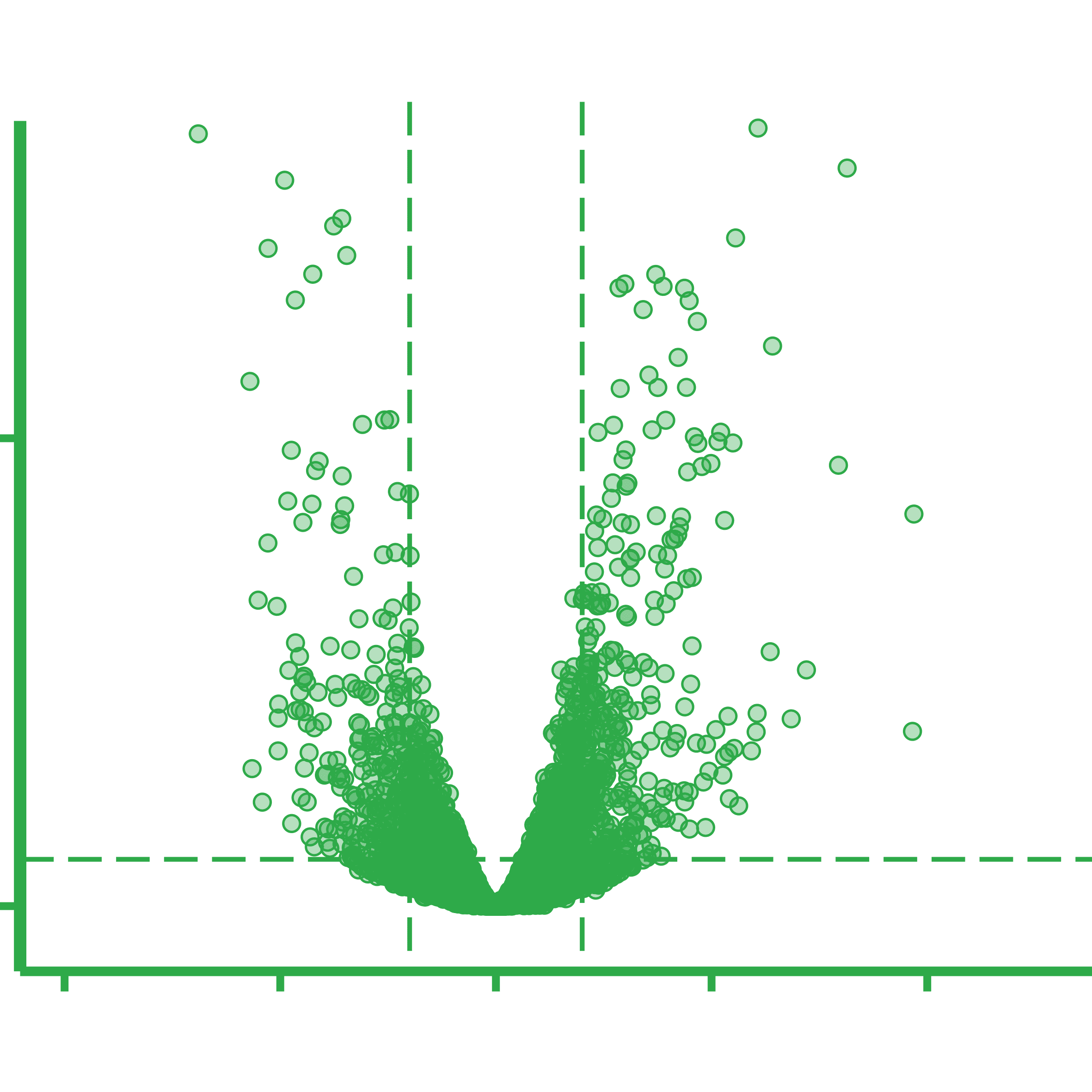
Overview of the general metatranscriptomics workflow. A) Sample total RNA is extracted from biomass, followed by B) ribosomal RNA (rRNA) depletion. C) cDNA sequencing libraries are prepared from the rRNA-depleted RNA and D) the resulting cDNA libraries are DNA sequenced. E) Obtained cDNA sequences are adaptor trimmed and quality filtered before F) depletion of residual rRNA reads bioinformatically. G) Filtered and depleted reads are mapped against a genomic reference, and the number of reads mapping per genomic feature is counted. H) Lastly, differential gene expression (DGE) analysis is done according to the relevant experimental design.
FAQ
Is Ribosomal RNA depletion necessary for my samples?
Ribosomal RNA constitutes up > 80% of cellular RNA. Our recommendation is to perform rRNA depletion for RNA high yield samples, to maximize focus RNA in the subsequent sequencing. For RNA low-yield, for which RNA-depletion due to recovery loss is not a recommended option, increasing sequencing depth may be used as an alternative solution.
How many replicates are needed to perform DGE analysis?
Statistical power increases with increasing number of replicates, especially for low-abundant genes. Our recommendation is a minimum 4 biological replicates per condition.
How many reads do I need?
Depends on the scientific question at hand and sample matrix. For DEGs 2-5 million mapped reads for pure cultures is sufficient for most prokaryotes, for whole transcriptome sequencing 100-200 mio reads is typical.
How should I collect samples for transcriptomic/metatranscriptomic profiling?
The transcriptome/metatranscriptome is prone to mRNA degradation and changes in expression profile. Hence, to minimize biases associated with sampling, special care must be taken. We recommend adding a mRNA preservative (preferably RNAlater) and snap freezing samples as soon as possible.